马上注册,结交更多好友,下载更多分子模拟资源。
您需要 登录 才可以下载或查看,没有帐号?我想注册


x
本帖最后由 小萌物 于 2018-2-4 11:26 编辑
2018-2-4更新:发现原来的代码存在小bug,已修改后上传到附件。目前最新版支持LeDock、AutoDock和Vina的对接盒子获取,兼容python2/3。 - Binding pocket
- xmin xmax
- ymin ymax
- zmin zmax
类似的,AutoDock和AutoDock Vina的盒子格式如下: - # Autodock Vina
- --center_x xx.x --center_y xx.x --center_z xx.x --size_x xx.x --size_y xx.x --size_z xx.x
-
- # AutoDock
- npts npX npY npZ # num. grid points in xyz
- spacing 0.375 # spacing (A)
- gridcenter CenterX, CenterY, CenterZ # xyz-coordinates or auto
3. 根据活性空腔的氨基酸确定盒子
LePro可以识别含有一个配体的蛋白活性空腔,但无法识别含多个小分子或离子或无配体蛋白活性空腔,如1MQ4。另外,由于没有图形界面,无法显示和调节盒子的位置。因此有必要用其他方法来获得盒子信息。论坛里eming用VMD和PyMOL Autodock Plugin分别实现了LeDock盒子信息的获取与显示 http://bioms.org/thread-1226-1-1.html。本来eming准备再写一个在PyMOL下获取盒子的教程,^_^我说我正好也在写这个代码,大神就把这个任务交给我了,感谢eming给我这次锻炼的机会,呵呵,做的不好的地方还请各位大虾指正。
首先介绍在PyMOL下获取盒子的原理和相应PyMOL Script代码的实现。对于不想看原理的伙伴们可以直接跳到下面看“用PyMOL Script编的PyMOL插件——GetBox Plugin”的安装和具体用法,基于这个插件可以在PyMOL中获取LeDock和Autodock Vina的盒子信息。 1. 预处理蛋白
首先是去除溶剂分子和离子。防止干扰后面的操作。 - cmd.remove('solvent') # 移除溶剂
- removeions() #调用移除离子函数
移除离子的代码: - cmd.select("Ions", "((resn PO4) | (resn SO4) | (resn ZN) | (resn CA) | (resn MG) | (resn CL)) & hetatm") #这里还不完善
- cmd.remove("Ions")
2. 根据配体确定盒子
这个方法用于含有配体的蛋白,选定蛋白的A链中小分子突出显示,其中若含多个分子,则手动选择配体,以配体盒子(ligand box)几何中心为盒子中心,生成对接盒子(docking box,图 1)。下面是关键代码: - cmd.select("ChaHet","hetatm & chain A") # 选中A链中小分子
- cmd.show("sticks", "ChaHet") # 以stick模式显示小分子,以便于手动选定配体getbox("ChaHet",extending) # 以配体几何中心为盒子中心,生成盒子,extending是指将配体盒子延长的大小
根据配体确定盒子的示意图

图 1. 根据配体确定盒子的示意图,以3CL0为例
选定对象盒子空间位置和大小信息的获取代码(关键):
- ([minX, minY, minZ],[maxX, maxY, maxZ]) = cmd.get_extent(selection) # 获得选定对象的盒子(ligand box),空间两点确定一个长方体
- minX = minX - float(extending) # extending是指docking box相对于ligand box,在minX方向延长的长度,默认值为5埃
- minY = minY - float(extending)
- minZ = minZ - float(extending)
- maxX = maxX + float(extending)
- maxY = maxY + float(extending)
- maxZ = maxZ + float(extending)
3. 根据活性空腔的氨基酸确定盒子
这个方法可用于没有配体的蛋白质活性空腔的确定。选定空腔各方向的氨基酸(>=2),以氨基酸们盒子(residues box)的几何中心为盒子中心,生成对接盒子(docking box, 图 2)。注意:氨基酸一般选择文献报导的活性空腔的,如果没有文献报道的,就最好用活性空腔搜索软件来确定,如CASTp、PASS、Pocket-Finder、PocketPicker等。下面是关键代码: - cmd.select("sele", ResiduesStr + " & chain A") # 选定链A中ResiduesStr中出现的氨基酸
- getbox("sele", extending) # 以氨基酸们的几何中心为盒子中心,生成盒子,原理与图1类似,但这里要注意extending大小的设置,默认值为5埃
根据文献报道的空腔氨基酸确定盒子的示意图

图 2. 根据文献报道的空腔氨基酸确定盒子的示意图,以3CL0为例
基于以上原理和方法,用PyMOL Script编了一个PyMOL的插件——GetBox Plugin,可以输出LeDock和Autodock Vina的盒子信息。首先介绍安装方法(图 3):打开PyMOL->Plugin->Install Plugin->打开下面附件中的GetBox Plugin.py->重启PyMOL->安装成功,PyMOL的Pllugin工具栏会多出一个菜单项GetBox Plugin,有三个子菜单,分别为:Advanced usage、Autodetect box、Get box from selection (sele)。
GetBox Plugin安装步骤

图 3. GetBox Plugin 安装步骤
Autodetect box的功能是打开蛋白后一键自动获取盒子,相应代码为autobox 5.0,适用于A链中只有一个配体的蛋白分析。
Get box from selection (sele)的功能是在选定了配体或氨基酸后一键获取盒子,相应代码为getbox (sele), 5.0,适用于含有配体的蛋白分析,也适用于没有配体但有文献报道的蛋白。
Advanced usage是“高级用法”的介绍,是针对以上两种方法参数固定的缺陷而设计的,使用者可以用GetBox Plugin自带的函数灵活地进行盒子分析,参数设定请看原理,这些函数包括: - * autobox [extending] (NOTES: solvent & some anions will be removed)
- this function autodetects box in chain A with one click of mouse, but sometimes it fails for too much ligands or no ligand
- e.g. autobox
- * getbox [selection = (sele), [extending = 5.0]]
- this function creates a box that around the selected objects (residues or ligands or HOH or others). Selecting ligands or residues in the active cavity reported in papers is recommended
- e.g. getbox
- e.g. getbox (sele), 6.0
- * resibox [Residues String, [extending = 5.0]]
- this function creates a box that arroud the input residues in chain A. Selecting residues in the active cavity reported in papers is recommended
- e.g. resibox resi 214+226+245, 8.0
- e.g. resibox resi 234 + resn HEM, 6.0
- * showbox [minX, maxX, minY, maxY, minZ, maxZ]
- this function creates a box based on the input axis, used to visualize box or amend box coordinate
- e.g. showbox 2,3,4,5,6,7
下面以3CL0为例,说明用法:法1:用PyMOL打开蛋白后,单击Autodetect box菜单项即可实现盒子识别,效果如图4和图5;
法2:在PyMOL命令窗口输入autobox 5.0可实现同样效果,若把默认值5.0改为其他数值,可以调节盒子的大小,如autobox 8.0;
法3: 选择配体Oseltamivir后,单击Get box from selection (sele)菜单可实现相同效果;
法4:选择配体Oseltamivir后,在PyMOL命令窗口输入getbox或者getbox (sele), 5.0可实现同样效果,可以调节盒子的大小,如getbox (sele), 8.0;
法5:查阅文献可以知道这活性空腔的氨基酸编号,在PyMOL命令窗口输入resibox resi 151+274+371, 5.0可实现类似效果。
另外,可以通过showbox函数 绘制box或调整box位置和大小。从图2中,可以看出, 有一小部分空穴没有包在box里,需要增大MaxY,减小MinZ。下图中盒子代码为showbox -40.4 , -23.2,-65.0 ,-47.5,0.8, 15.4,在PyMOL命令窗口输入showbox -40.4 ,-23.2,-65.0 ,-46.5,-0.5, 15.4可实现Y和Z方向的改变。
ps. 在PyMOL中配体的选择有很多种方法,例如:1. 打开蛋白序列窗口查看蛋白序列,一般配体在序列末端,点击即可选中;2. 打开蛋白质时会有配体信息,直接用select (sele),resn "配体缩写(一般为三个字符)" ,即可选中;3. 在图形窗口,点击All->A->present->ligand sites->cartoon即可显示配体;4. 采用GetBox Plugin 的Autodetect box菜单或autobox命令,即可选中配体分子(ChaHet)为球状模型,若隐藏其他lines、cartoon就很清晰。
如不懂参数设定,请看原理部分
Autodetect box获得3CL0的盒子

图 4. 3CL0盒子示意图
Autodetect box获得3CL0的盒子

图 5. 3CL0盒子对活性位点氨基酸包合情况示意图
在PyMOL的输出窗口中生成的盒子信息: - *********AutoDock Vina Binding pocket*********
- --center_x -31.8 --center_y -56.2 --center_z 8.1 --size_x 17.2 --size_y 17.5 --size_z 14.6
-
- *********AutoDock Grid Option*********
- npts 45 46 38 # num. grid points in xyz
- spacing 0.375 # spacing (A)
- gridcenter -31.800 -56.250 8.100 # xyz-coordinates or auto
-
- *********LeDock Binding pocket*********
- Binding pocket
- -40.4 -23.2
- -65.0 -47.5
- 0.8 15.4
- BoxCode(box_3387) = showbox -40.4, -23.2, -65.0, -47.5, 0.8, 15.4
GetBox PyMOL Plugin下载地址: |



 窥视卡
窥视卡 雷达卡
雷达卡 发表于 2014-7-30 19:48:37
发表于 2014-7-30 19:48:37

 提升卡
提升卡 置顶卡
置顶卡 沉默卡
沉默卡 喧嚣卡
喧嚣卡 变色卡
变色卡 千斤顶
千斤顶 显身卡
显身卡 楼主
楼主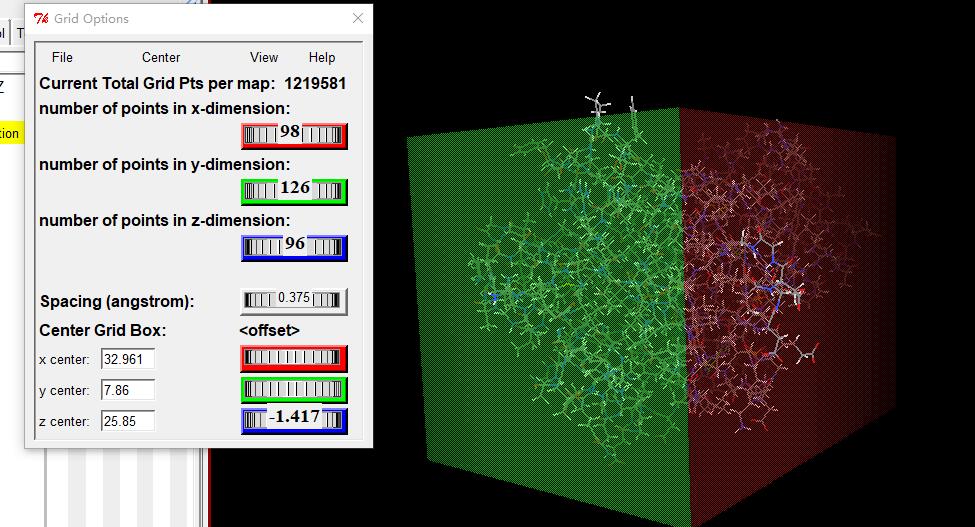

 发表于 2015-6-30 19:00:57
发表于 2015-6-30 19:00:57

