马上注册,结交更多好友,下载更多分子模拟资源。
您需要 登录 才可以下载或查看,没有帐号?我想注册


x
手把手教你怎么做虚拟高通量筛选,包括结果分析步骤及分析脚本。
1. 前言
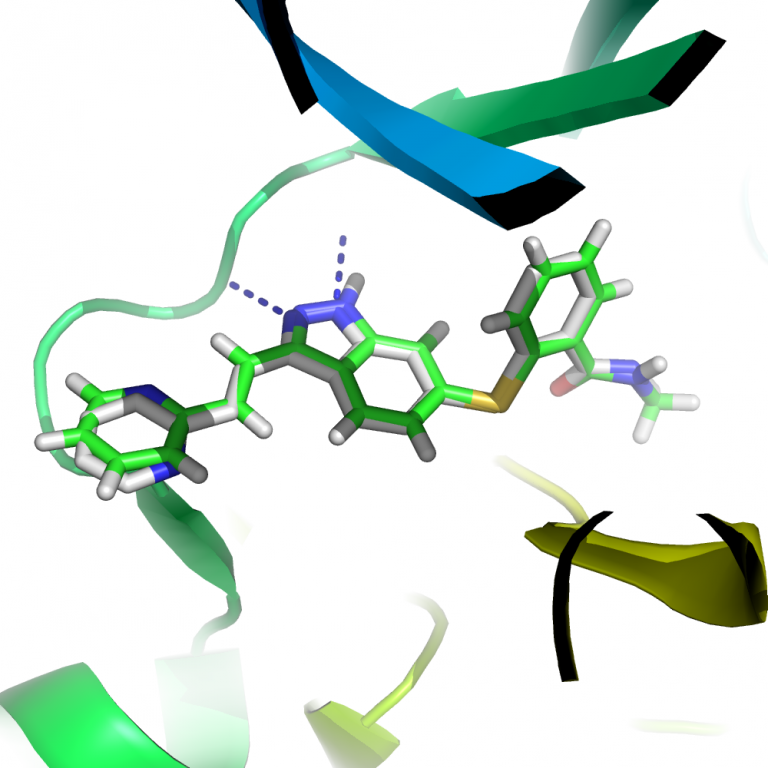
药物设计中活性化合物的发现可谓是大海捞针,西方谚语云“a needle on a haystack”。神农氏尝百草的原始方法已然不能满足现代药物开发的需求。即使基于现代自动化技术,动辄数十万乃至上百万的化合物筛选依然耗资数百万美元,历时一年左右。计算机辅助药物设计——在分子层面上理解药物分子与靶点的作用,则提供了一种快速、低廉的虚拟筛选方案,在先导药物发现中效果显著。 血管内皮生长因子(vascular endothelial growth factors,VEGFs)及其受体(vascular endothelial growth factor receptors,VEGFRs)在肿瘤血管的新生中起重要作用。特别是VEGF所介导的VEGFR-2信号通路,促进了一些新血管形成所需的内皮细胞响应,如细胞增殖、迁移和存活。由于VEGF/VEGFR-2信号通路在肿瘤血管的生成中起关键作用,阻断VEGF/VEGFR-2信号通路已经成为治疗癌症的一种有吸引力的途径。目前已有六个可有效抑制VEGFR-2的多靶点小分子酪氨酸激酶抑制剂上市,用于治疗肾癌、肝癌、胃肠道间质瘤等各种癌症。辉瑞(Pfizer)公司的Axitinib(商品名称Inlyta)2012年获得FDA批准用于肾癌的治疗。除抑制VEFGR2之外,最近的研究表明Axitinib也可有效抑制BCR-ABL1基因的T315I突变。ABL1激酶是癌症治疗领域明星药物格列卫(Gleevec,诺华)的靶点,不过Gleevec对患有T315I突变的病人无效。 本文针对VEGFR2激酶,采用计算机辅助药物筛选方法,对美国国家癌症研究所(NCI)的多样化小分子化合物库进行筛选,旨在为对计算机辅助药物设计感兴趣的研究人员提供一个可参考的筛选方案,具体计算涉及:蛋白质受体准备、高通量对接、对接结果分析、基于经验(Knowledge-based)的化合物筛选准则等。附件包含了该项工作所需文件以及分析脚本。计算软件如LeDock、LeFrag、LeWater以及LePro可从LePhar Research官网(http://lephar.com)上下载,目前试用版本有效期至2015年11月份。计算平台为Linux或Mac OS。 2. 计算方法 从Protein Data Bank(PDB,http://www.rcsb.org)下载VEGFR2与Axitinib的复合物晶体结构(PDB code 4AG8)。附件目录receptor下已经包含了该复合物结构。可以直接下载本文附件,按照文中介绍的操作流程进行练习。打开终端(Terminal),进入附件中的work目录,文中所有操作均在work目录下进行。首先要对该蛋白结构进行预处理,以得到一个可用于分子对接的蛋白结构。具体的处理步骤包括:1)去水、配体、离子等;2)加氢。用LePro软件进行自动处理 $ lepro_linux_x86 ../receptor/4AG8.pdb 处理后的蛋白文件名为pro.pdb。该步骤同时生成dock.in文件,该文件是分子对接的参数文件。分子对接的一个极其重要参数是结合位点的设置。默认的结合位点系在Axitinib的基础上向四周延伸4 Å。 从ZINC(http://zinc.docking.org)下载NCI Diversity 3小分子数据库,选择pH 6-8的Mol2文件下载。该文件位于nci_diversity目录中。NCI多样化小分子数据库含1597个化合物,这些化合物可从NCI免费获得。ZINC根据分子对接的要求,对这些化合物进行了预处理,对应生理pH值生成各种可能的同分异构体。因而,下载文件中包含的化合物实际为2044。把这个小分子库分割成单个的化合物分子文件 $ lefrag_linux_x86 -spli ../nci_diversity/ncidiv.mol2 该步骤把ncidiv.mol2中包含的2044个分子分割成2044个小分子文件,每个小分子文件名以ZINC开头。 $ cp ../receptor/4ag8_lig.mol2 . $ ls *mol2 >ligands 把Axitinib(文件名为4ag8_lig.mol2)也加到筛选数据库中,作为衡量计算性能的内在参考。上述第二个命令则生成虚拟高通量筛选的化合物列表。 $ ledock dock.in & 对接完全部2044个分子大约需要一天时间。 3. 结果分析 可以等对接全部完成,或者在对接进行过程中进行如下分析。 $ ../scripts/anal.csh 结果保存在文件docking_summary.txt中。我们可以用以下命令查看该文件 $ less docking_summary.txt 第一列为配体文件名,第二列为结合能,第三列为配体结合效率(结合能除以配体的重原子个数)。按下Q字符键退出查看。 $ ../scripts/selection.csh 首先以结合能-7 kcal/mol作为阈值。虚拟筛选在实践中的一个显著问题是假阳性率太高,很多预测有活性的化合物实际上是没有活性的,这是由计算的近似性决定的,也跟配体结合蛋白时候复杂的热力学、动力学性质相关。针对计算的内在特性,可以考虑用配体结合效率(LE)进行排序。通常衡量先导化合物的标准是配体结合效率(LE)不小于0.3。实践上,通常同时以结合能以及配体结合效率(LE)来控制假阳性率。譬如以结合能-7 kcal/mol以及配体结合效率0.3作为阈值。 激酶抑制剂一般与Hinge区域形成两个氢键(见下图中的蓝色虚线),可用此经验规则进一步控制假阳性率。用LeWater来批量自动检测每个配体的第一个对接构象与蛋白靶点的氢键作用情况(具体实践中往往要考虑所有可能的对接构象,而不仅仅是第一个对接构象!),然后要求每个小分子与蛋白结合时必须与Hinge形成两个氢键。LeWater根据输参数文件(scripts目录中的lewater.in)中的输入自动批量检测每个构象与蛋白的氢键作用情况,并对每个氢键根据其几何构型进行打分,分值从0到1——0表示没有氢键形成,1表示氢键最理想。脚本中用0.5作为阈值。 4. 讨论 LeDock预测Axitinib与VEGFR2的结合能为-11.8 kcal/mol。实验测得的结合能为-12.2 kcal/mol,可见LeDock能够很好地预测化合物的结合能。同时,LeDock预测的结合构象与晶体结构中的基本完全一致。 参考文献
| 

 窥视卡
窥视卡 雷达卡
雷达卡 发表于 2015-5-17 18:31:20
发表于 2015-5-17 18:31:20

 提升卡
提升卡 置顶卡
置顶卡 沉默卡
沉默卡 喧嚣卡
喧嚣卡 变色卡
变色卡 千斤顶
千斤顶 显身卡
显身卡
